
Technology
Our novel approach relies on Pre-Templated Instant Partitions (PIPs) to simultaneously segregate complex cell mixtures into partitions with barcoded template particles that can be easily processed for single cell applications such as single cell RNA sequencing (scRNA-Seq). This approach (PIPseq™) eliminates the need for complex, expensive instrumentation and microfluidic consumables.
Benefits of Fluent’s PIPseq V Technology for Gene Expression Studies
Better Sensitivity Provides More Usable Data Across More Sample Inputs
- Improvements in the synthesis, amplification, and purification of the captured targets produce more genes & transcripts per cell
- TruIDTM introduces a completely novel way to count target molecules that removes bias and sequencing artifacts caused by randomer bar codes
- Wash steps in our process drastically reduce the levels of ambient RNA background
- PIPseq’s scalable PIP partitioning system generates far fewer multiplets than other methods which means more usable data
Fluent BioSciences PIPseq Single Cell Workflow
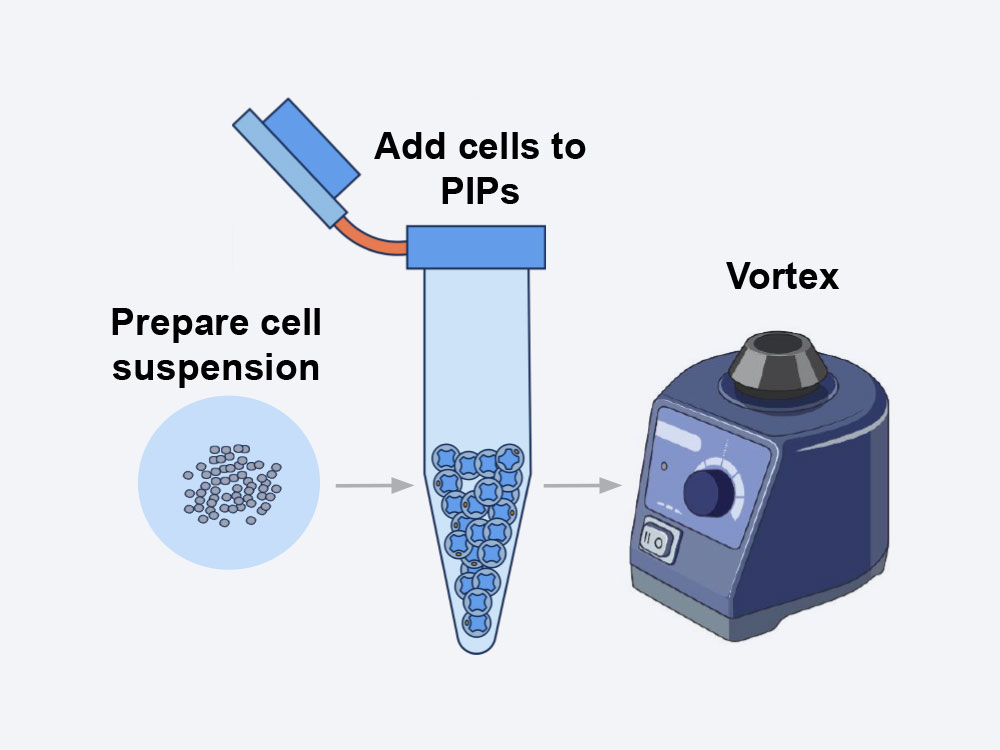
1. Sample Preparation
- During sample preparation using the PIPseq V SCRNAseq kit, the cell suspension of interest is mixed with our core template particles and segregated into Particle-Templated Instant Partitions (PIPs) by simple vortexing
- The cells in PIPs are then lysed on a thermal device and the mRNA is captured by barcoded oligonucleotides incorporated with the template particles
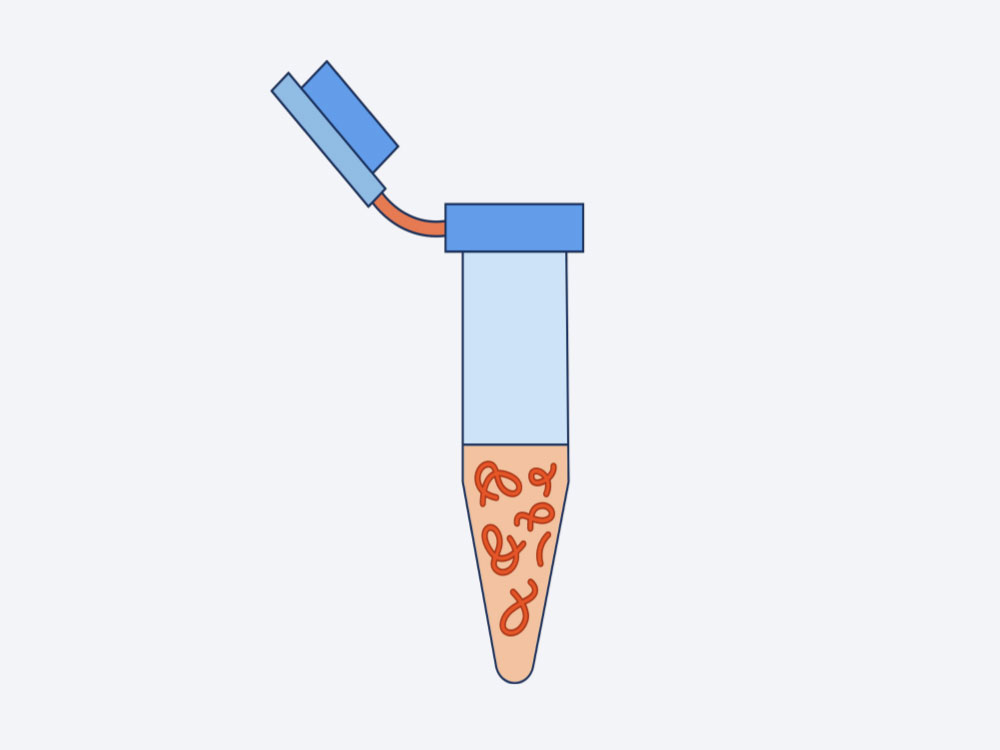
2. Library Preparation
- The captured mRNA undergoes reverse transcription and cDNA libraries are then generated via fragmentation, end repair and A-tailing
- This is then followed by adapter ligation and cleanup
- Sample indices (compatible with short-read sequencing) are then added, followed by a final cleanup

3. Next generation sequencing (NGS)
- Libraries can then be processed on the appropriate Illumina NGS instrument.
- The sequencing reagent kit (and chip) is chosen based on the number of samples multiplexed, cell count and desired read depth

4. Data Analysis
- The Fluent PIPseekerTM Software enables primary analysis of the PIPseq sequencing libraries
- Upload your FASTQ files into the software and obtain summary metrics, diagnostic plots, clustering and differential gene expression tables
- Also generate standard feature-barcode count matrices, which are compatible with widely-used open-source tertiary analysis packages
Related Resources
Select from the below resources to learn more about our protocols (user guides) or customer applications (posters).